And where are we going next?
This graph never ceases to amaze me. On the horizontal axis we have dates from September 2001 to July 2015. On the vertical axis we have the cost to sequence a million base pairs of DNA, with the axis having a
logarithmic scale (each tick mark is multiplied by 10, e.g. change from 10 to 100 to 1000). The blue line describes what is called
Moore's Law which describes the increase in purchasing power as computer costs come down. The rate of improvement in DNA sequencing easily outpaces the improvement in computing. Since September 2001, the price of DNA sequencing has dropped 6
orders of magnitude from $5,292.39 to $0.015. From more than $5,000 to less than 2 cents!!!
 |
Same data as above by with the vertical cost axis
on a normal scale, not logarithmic. |
What caused the drop in sequencing from April 2015 to July 2015? This is due to the release of the
Illumina HiSeq X Ten system. Previously this system was only licensed for human sequencing. But on
October 6th, this restriction was lifted and this system is now available for livestock genomics (and other applications). This means we can now sequence cattle genomes for a little more than a $1,000.
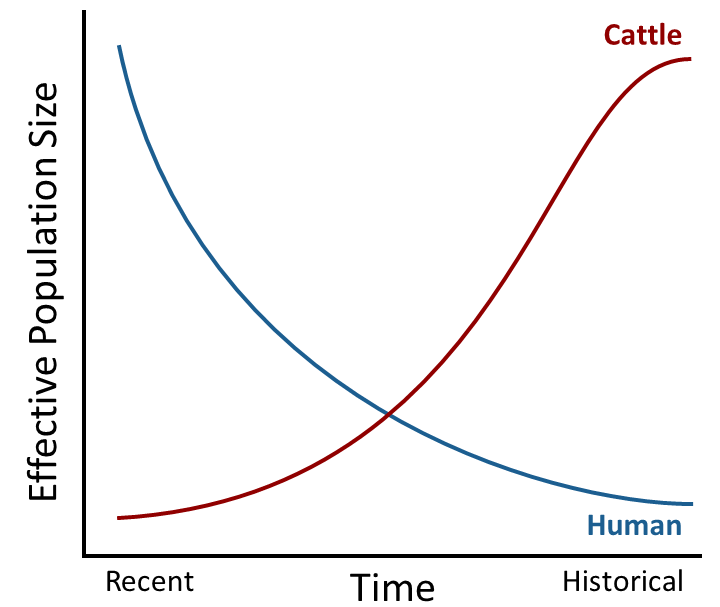
What do we plan to do with this decreased sequencing cost? Sequence a lot of bulls and cows of course! As we started to look at this sequence data, what have we found? First of all, we find a lot of DNA variants. In fact, we find similar numbers of variants to when we sequence human genomes. But in cattle we find many common variants (at a moderate frequency in the population) whereas in humans we find many rare variants (very low frequency in the population). This is because humans had a very small number of parents in the past (small
effective population size) and a growing number of parents since that time (increasing effective population size).
 |
Cartoon of changes in population structure of humans
and cattle. |
Cattle have the opposite pattern. The wild ancestors of cattle (
aurochs) had a very large population in the past with a large number of parents (large effective population size), and through the processes of domestication, selection, and breed formation the effective population size has decreased. Think about how many times you find the same popular bulls in your cattle's pedigrees; this makes the effective population size smaller. This small effective population size with a large number of common variants is what makes downstream uses, like genomic-enhanced EPDs, of genomic data so effective in cattle.
So what are the downstream analyses of genomic data? Well there are lots, too many to mention in this post. But one of the first techniques we use in livestock genomics is a process called imputation. I know imputation is another one of those fancy jargon words, but it simply means that we can infer the genotypes at untested DNA variants based on the patterns of tested DNA variants. Now, instead of running our analyses with tens of thousands of DNA variants spread across the DNA like "mile markers", we can analyze millions of variants and get closer to our "points of interest". Using millions of variants we find important regions that we missed in earlier analyses and more precisely identify the regions of DNA that contain the important genes and causal variants that influence our production traits.
Using genomic sequencing data, scientists have identified causal variants for things like genetic defects, variants responsible for pregnancy losses (embryonic lethals), coat color, and horned vs polled. But finding these causal variants is not easy. Rather than being a simple base pair substitution (SNP), these causal variants are frequently structural variants, meaning large chunks of DNA are inserted, deleted, or rearranged.
As we generate more and more whole genome sequence data, we will have new SNP tests (a.k.a. SNP chips or SNP assays) developed. But instead of these SNPs (DNA variants) being evenly spaced like mile markers, they will be concentrated in genes and other functionally important regions. We will see SNP tests that genotype hundreds of thousands of SNPs, which will be mostly used in research settings. The valuable or interesting variants from these research studies can then be put on low density SNP tests. These low density SNP tests will then be more valuable and predictive as they will be focused on functional content, the points of interest.
Another interesting idea is for breed associations to develop programs to sequence the entire DNA of important or popular sires (idea originated with Jerry Taylor). I call this genomic "surveillance." For example, members would pay an extra $1 every time they registered a calf. When a bull has 1,500 registered progeny, he has now earned $1,500 in fees, and the breed association has enough money to sequence his entire genome. This allows breed associations to discover and keep track of new DNA variants that are rising to high frequency in their population.





Comments